
Retinitis Pigmentosa (RP) Surgical Programs
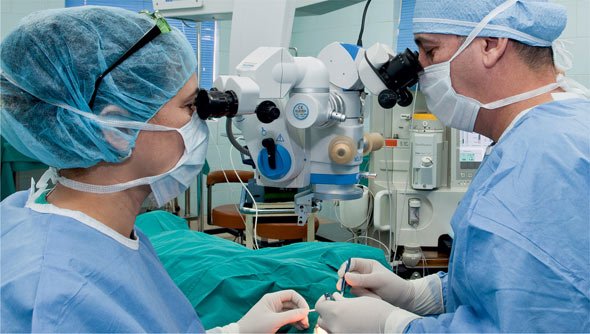
Retinitis Pigmentosa refers to a number of inherited eye diseases (including rod-cone dystrophy, usher’s syndrome, leber’s amaurosis and batten’s disease) that causes retinal degeneration leading to a slow progressive vision loss. This condition is due to damage in the rod and cone photoreceptors. Rod photoreceptors are the retinal cells responsible for peripheral vision and night vision and function best in dim light while cone photoreceptors are the retinal cells responsible for color vision and color sensitivity and work best in bright light. With PR the rods photoreceptors usually first disintegrate making the loss of night vision the first symptom. The breakdown in the rod photoreceptors brings about a breakdown in the cone photoreceptors. On rare occasions, the cone photoreceptors are first affected bringing about an interruption in central vision and color perception. With time, damage to the cone cells causes damage to the rod cells leading to tunnel vision and loss of night vision.
PR affects about 1 in 3500 people and as many as 6% of deaf people. The most prevalent theory contributes the development of Retinitis Pigmentosa to genetics (though it might skip a generation or more before showing up). Symptoms usually appear in childhood with serious vision loss normally occurs in early adulthood progressing throughout one’s life.
Symptoms will include:
- Problems distinguishing colors
- Problems in reading
- Problems in seeing details
- Problems in seeing in dim light
- The loss of night vision
- The loss of side vision
TREATMENT
Cuba offers specialized Retinitis Pigmentosa (PR) eye programs which include ophthalmologic medical examination: biomicroscopy, tonometry, fundus, Visual acuity, refraction, electrophysiological studies, perimetry, optical coherence tomography.
- Retinitis Pigmentosa treatment. First visit (21 days hospitalization)
- Retinitis Pigmentosa treatment. Second visit (14 days hospitalization))
- Retinitis Pigmentosa treatment. Third visit (14 days hospitalization)
PLEASE NOTE THE FOLLOWING IMPORTANT INFORMATION:
- The above program does not include medications for certain conditions such as depression, blood coagulation, etc.
- Medicaments, fluids, blood and derivatives to be used, as well as additional procedure(s) performed not included in the exact treatment would be invoiced separately on upon the conclusion of said treatment / procedure(s)
ACCOMMODATION:
Private room with the following features:
- Electronic patient bed
- Equipment for disabled patient
- Oxygen hookup
- Three à la carte meals taking into account the patient’s preferences and / or special diets prescribed by physician
- Fully equipped private bathroom
- Infirmary and nursing care
- Colour TV with national and international channels
- Local and international phone services (extra cost will apply)
- Safe box
- Internet service on every floor
- Laundry services
ADDITIONAL SERVICES INCLUDED IN THE PROGRAM:
- Assistance in visa issuance and extension (If needs be)
- Each patient/ companion will be assigned a multi-lingual field member with the mandate of attending to all of our patients’ translation and personal needs;
- 20 hours internet service;
- Local airport pickup and drop off; and
- Hospital pickup and drop off (if needed)